IonGAP has been designed with the aim of simplifying your work, and our Web interface has a lot to do with it. Creating a project in IonGAP is so simple as following the next steps:
Click here or use the Home tab of the navigation bar to go to the Home page. Then, click the Start New Project button under the platform description.

In the New Project form, you will see four sections:
- A first section where you must fill a few mandatory fields:
- Project name: a descriptive identifier for the project (usually, the organism name). It must be 3 to 50 characters long, containing only letters, numbers, underscores or hyphens. It is advisable to avoid using previous project names.
- Email address: this address will be used exclusively to notify you once the project has started, as well as once it is complete. It will not be used in any other way, nor transferred to anyone external to the service.
- Institution: the name of the institution you belong to will be used only with informative/statistical purposes. It must contain only letters, numbers, spaces, underscores or hyphens.
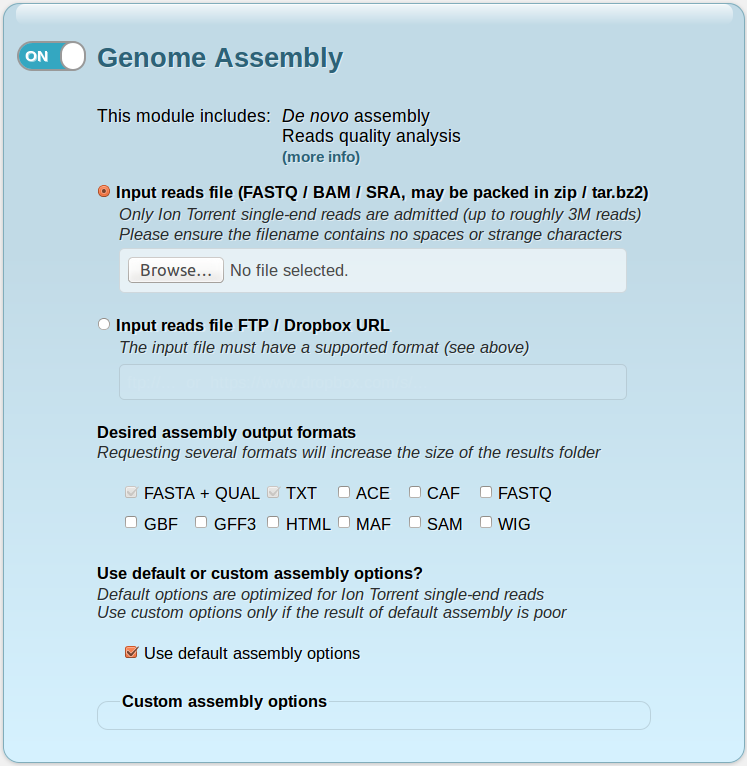
- A blue section corresponding to the Genome Assembly module of the service, which you can enable or disable by clicking on the switch in the upper left corner. When enabled, it contains the following fields:
- Input reads file: this is the only file needed to perform the assembly. It must have FASTQ (.fastq, .fq), BAM (.bam) or SRA (.sra) format, and its name cannot contain spaces or strange characters (such as slashes, asterisks, brackets or parentheses). IonGAP allows compressed files in zip or tar.bz2 format, which can be obtained directly from the Torrent Server's FileExporter plugin and are useful in order to reduce the upload time. Currently, IonGAP admits datasets of up to roughly 3 million Ion Torrent single-end reads. This corresponds to about 1 GB in FASTQ format (the size will vary largely depending on the input format). You can attach the file by clicking the Browse... button.
- Input reads file FTP / Dropbox URL: alternatively, you can supply a FTP or a shared Dropbox URL to your reads file (which must be in one of the admitted formats, see above). This helps to avoid long upload times, especially when using the same file in several projects. Please ensure that the URL follows the form indicated in this text field.
- Desired assembly output formats: here you can choose between 9 optional output file formats for the assembly module. The default formats are FASTA + QUAL and TXT.
- Use default assembly options: by unchecking this option, you can specify a custom assembly configuration, changing any of the 14 configurable assembly parameters. However, this is not advisable unless you are obtaining poor results using the default assembly configuration. The default parameter values come from an exhaustive comparative study on Ion Torrent single-end sequence data.
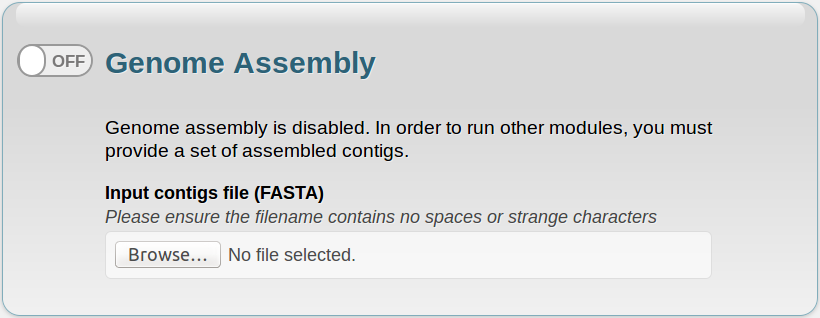
If you disable this module, a new panel will appear below, where you can submit a file containing your own assembled contigs. This file must be in FASTA (.fasta, .fna, .fas) format, and its name cannot contain spaces or strange characters (such as slashes, asterisks, brackets or parentheses).
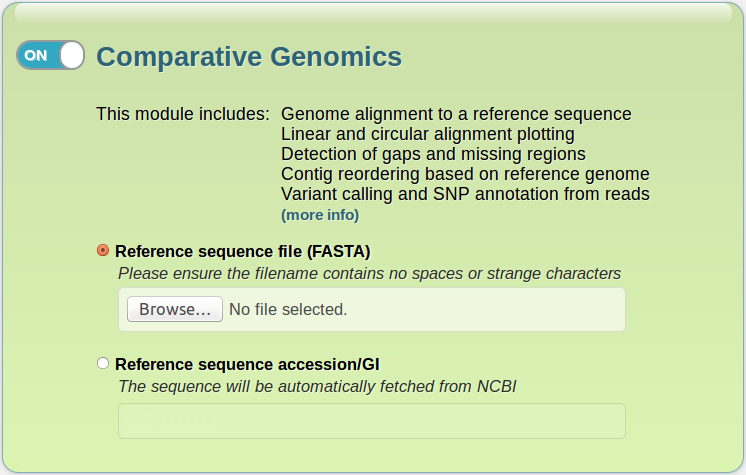
- A green section regarding to the Comparative Genomics module, which you can enable or disable by clicking on the switch in the upper left corner. It contains the following fields:
- Reference sequence file: in order to perform the comparative analysis routines, you must provide a reference sequence file. This file must be in FASTA (.fasta, .fna, .fas) format, and its name cannot contain spaces or strange characters (such as slashes, asterisks, brackets or parentheses).
- Reference sequence accesion/GI: if your reference sequence is in the NCBI database, you don't need to download it. Simply type its accesion or GI number here, and IonGAP will take care of the rest.
- An orange section corresponding to the Bacterial Classification and Annotation module of the service, which you can enable or disable by clicking on the switch in the upper left corner. It contains the following fields:
- Filter plasmid alignments: uncheck this option if you want to get the unfiltered output of the identification of plasmids. If checked, only the two best-scoring plasmid sequence hits for each alignment, having at least 500-bp length and 90% identity, will be included.
- Filter virulence factor alignments: uncheck this option if you want to get the unfiltered output of the identification of virulence factors. If checked, only the two best-scoring sequence hits for each alignment, having at least 90% identity, will be included.
- Perform MLST: check this option if you want MLST to be performed and you know the organism species (scheme). If checked, you must select the scheme from the pull-down list.

Once you have filled the form, click the Start project button. A green label will appear, telling you to wait while your files are transferred. This process can take from 5 minutes to an hour, depending on the data size and the quality of your connection. You can use your computer normally meanwhile.
After the files have been uploaded, you will see a new page informing that your project has been started. You will receive an email notification to the address you provided when your project starts, as well as another email once it is complete, through which you will be able to download the project results.