The IonGAP workflow consists of three stages or modules. However, the user is allowed to disable any of these modules if desired, in order to speed up the whole process. A test dataset is available for trying the platform.
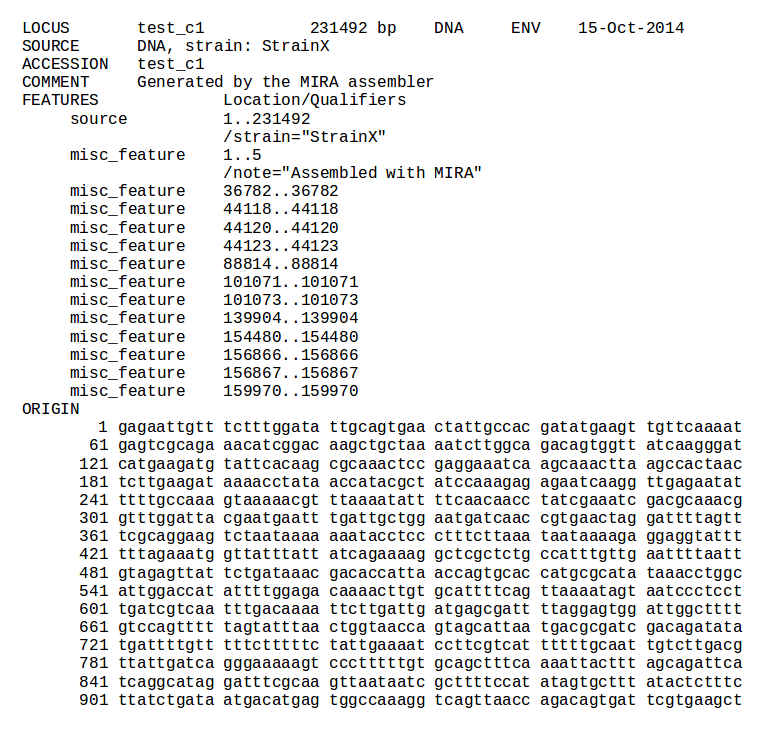
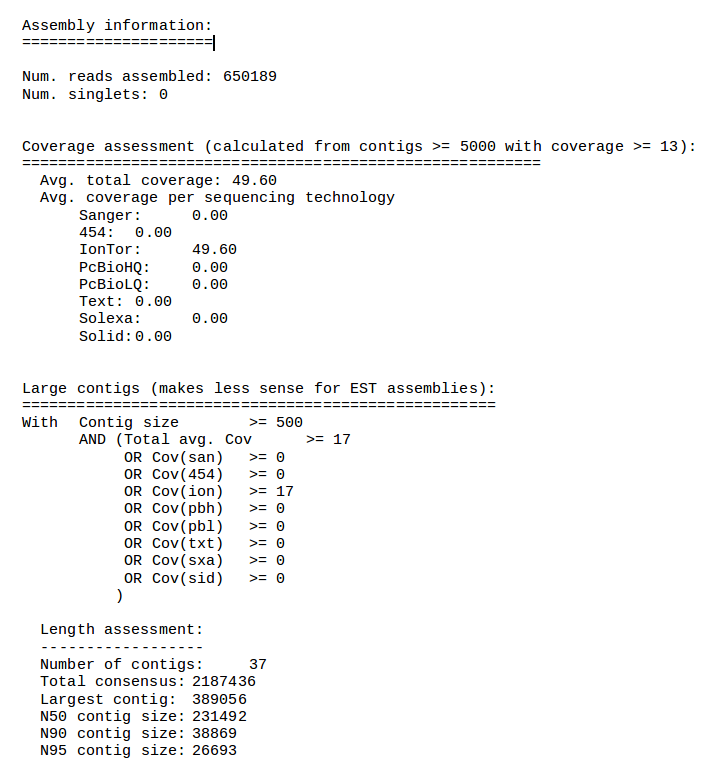
The Genome Assembly module is mainly composed by the MIRA assembler. The determination of the most suitable assembler for Ion Torrent data, as well as the assembler optimization, have entailed a series of exhaustive comparative studies. Moreover, the configuration of the assembler has been greatly simplified by means of a user-friendly Web interface, which allows to start an assembly project just by submitting a FASTQ or BAM file. This file may be compressed in zip or tar.bz2 format (the output compression formats offered by the Torrent Server's FileExporter plugin), in order to reduce the upload time. The user is allowed to configure a variety of relevant assembly parameters, in case the default assembly is not satisfactory, as well as to choose between 11 assembly output formats.
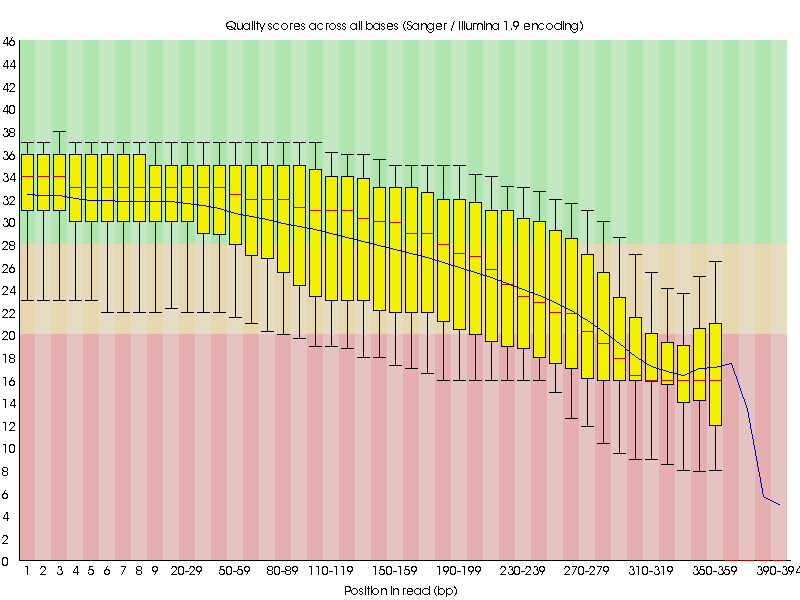
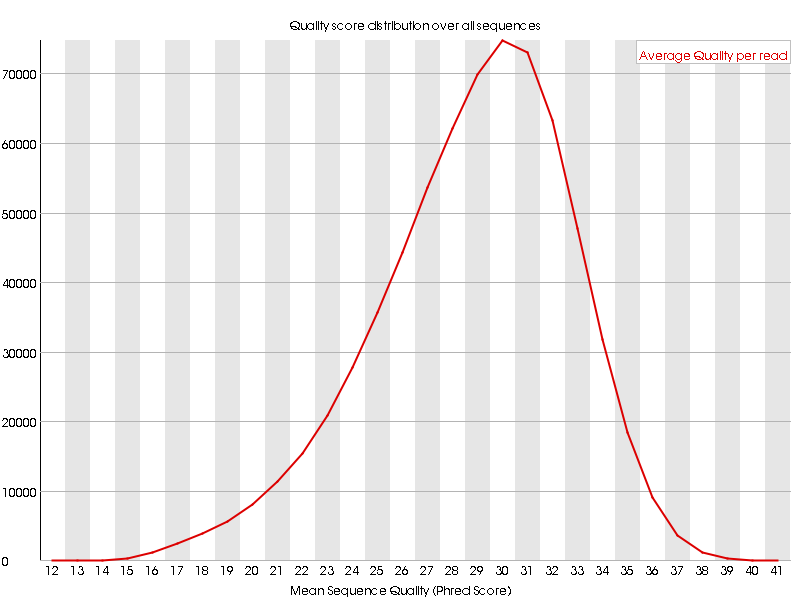
The assembler is set to deal with Ion Torrent single-end reads, which must be contained in a single file and will be also analyzed by FastQC in order to generate an informative quality report. Due to resource limitations, IonGAP allows readsets of up to about 3 million Ion Torrent single-end reads. This corresponds to about 1 GB in FASTQ format (the size will vary largely depending on the input format). We do not recommend preprocessing or trimming the reads before submitting, but users could find random sampling or digital normalization useful in order to reduce the amount of reads. The processes which compose this module and the results obtained from each one are detailed in the table below.
| Application | Process | Result | Result examples |
|---|---|---|---|
| MIRA | Genome assembly | Assembled contigs in various formats, assembly information and statistics | Examples |
| FastQC | Reads quality analysis | Quality analysis report | Examples |
The assembly stage may also be disregarded, as the service allows the user to supply a set of assembled contigs in FASTA format. This makes the rest of the pipeline accessible to users of other sequencing platforms.
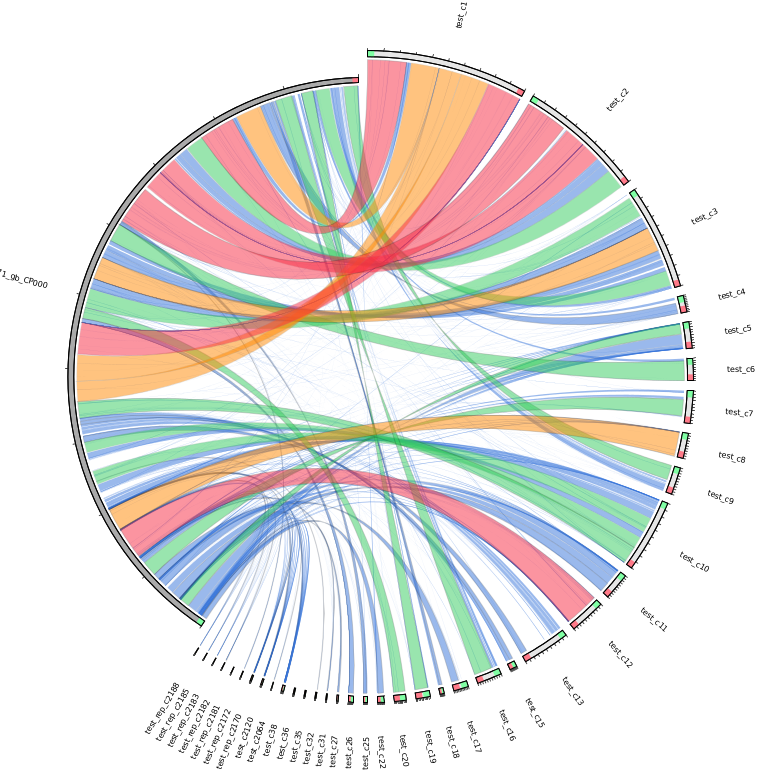
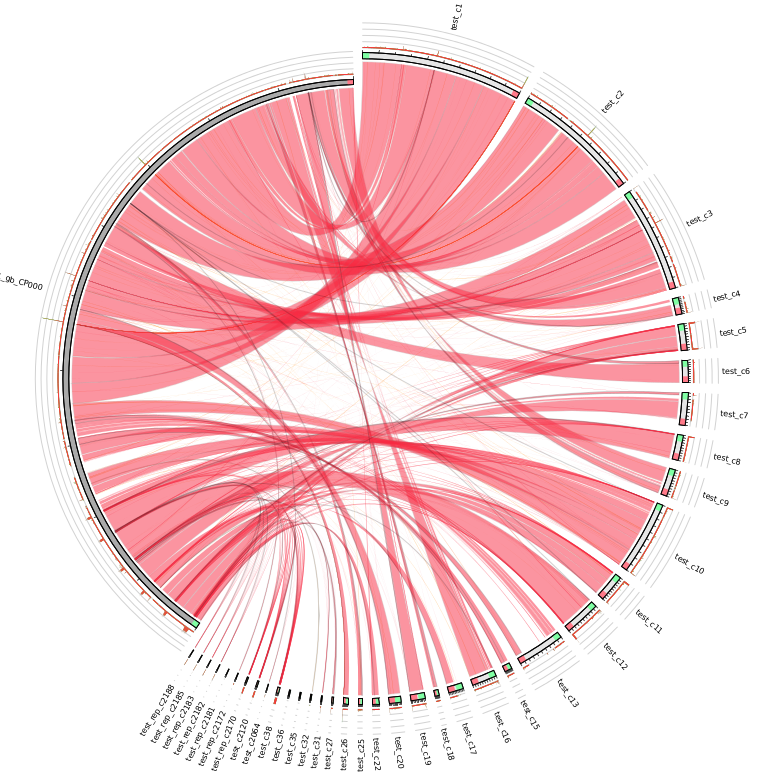
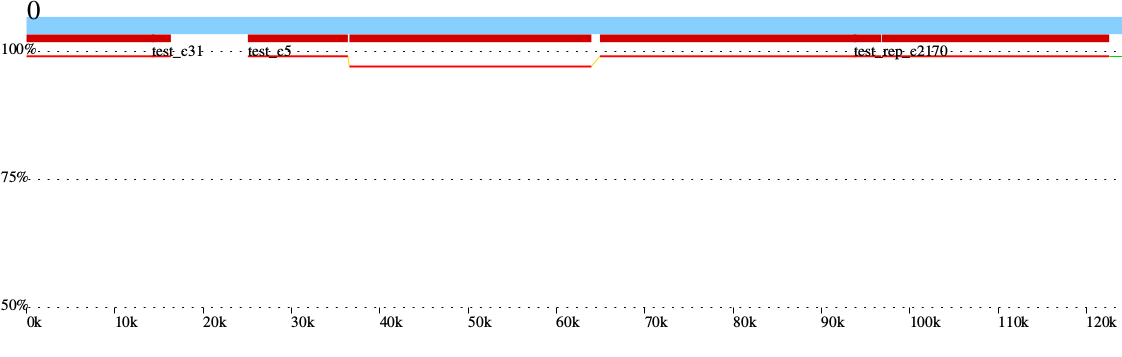
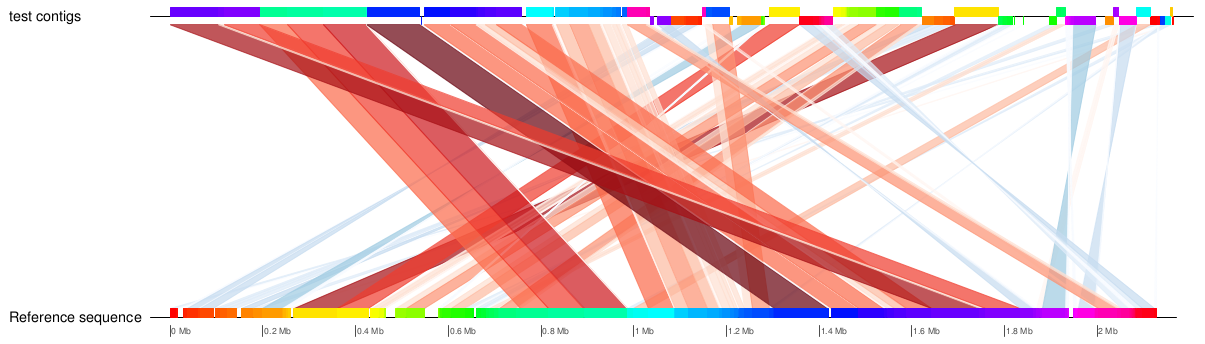
Following the assembly, if the user provides a reference sequence (or its NCBI accession/GI number), there is a succession of comparative analysis processes performed by different external applications. The result of this stage is a set of graphical and textual reports of the alignment between the contigs and the reference sequence, as well as relevant information derived from it, such as the set of gaps and missing regions found in the assembly. If a set of sequence reads is available (Genome Assembly module enabled), variant calling and annotation of SNPs are also performed. The tools involved in this stage and the results obtained from each one are detailed in the table below.
| Application | Process | Result | Result examples |
|---|---|---|---|
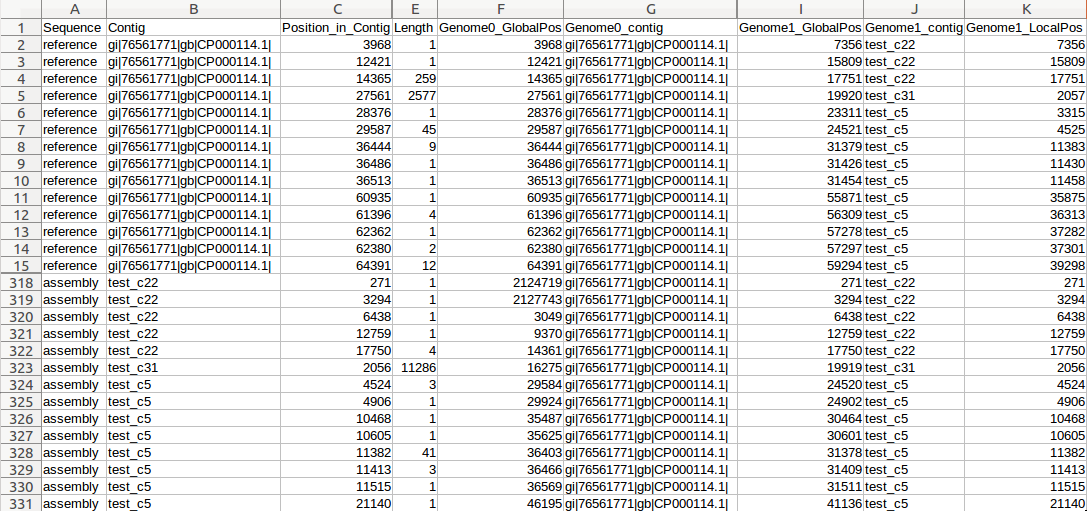
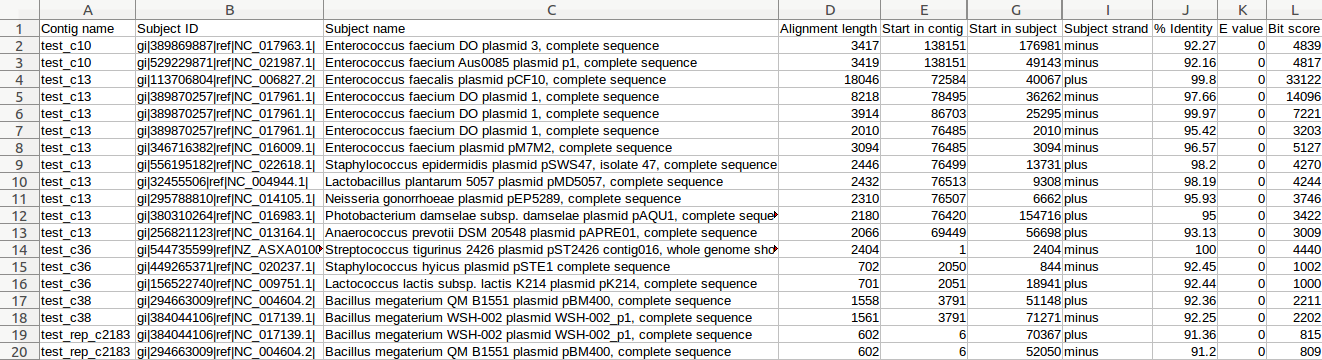
| MUMmer, Circos, Circoletto, genoPlotR | Genome alignment | Linear and circular alignment graphs | Examples |
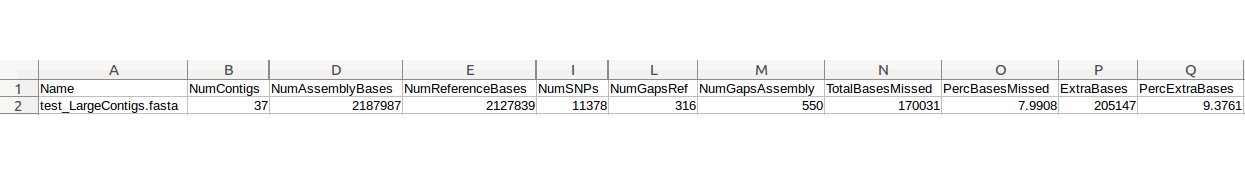
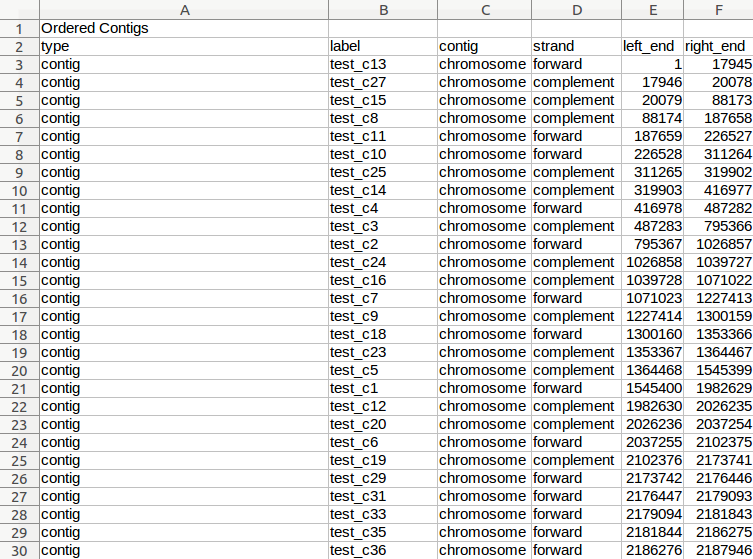
| Mauve | Genome alignment, contig reordering | Reordered contigs, alignment summary, information on gaps and missing regions of the assembly | Examples |
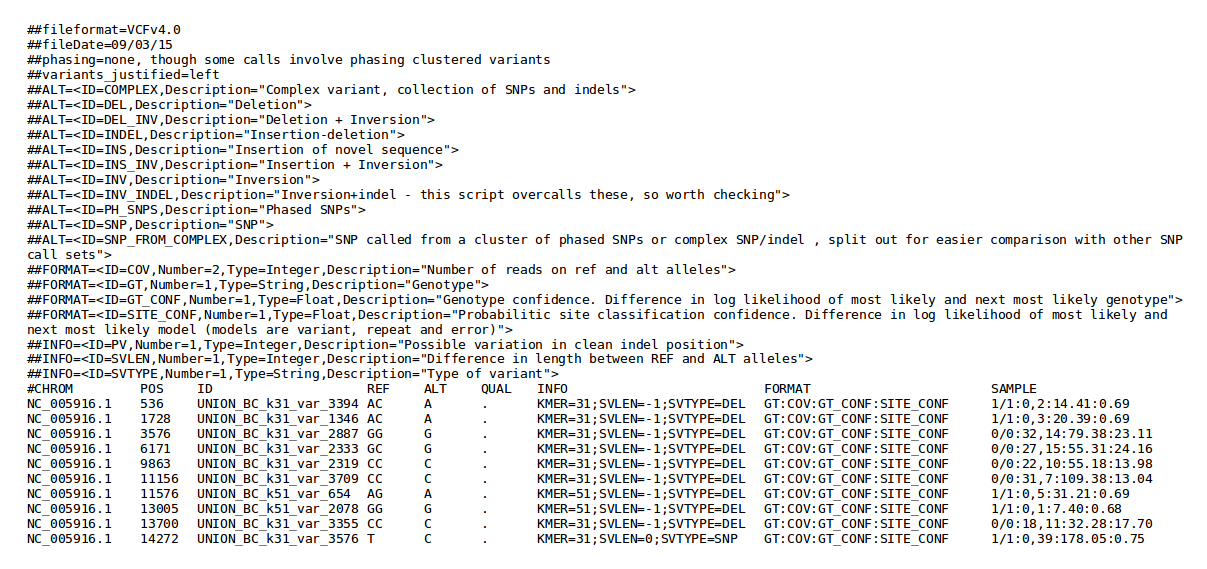
| Cortex | Variant calling | Variant calls (from raw reads) in VCF format | Examples |
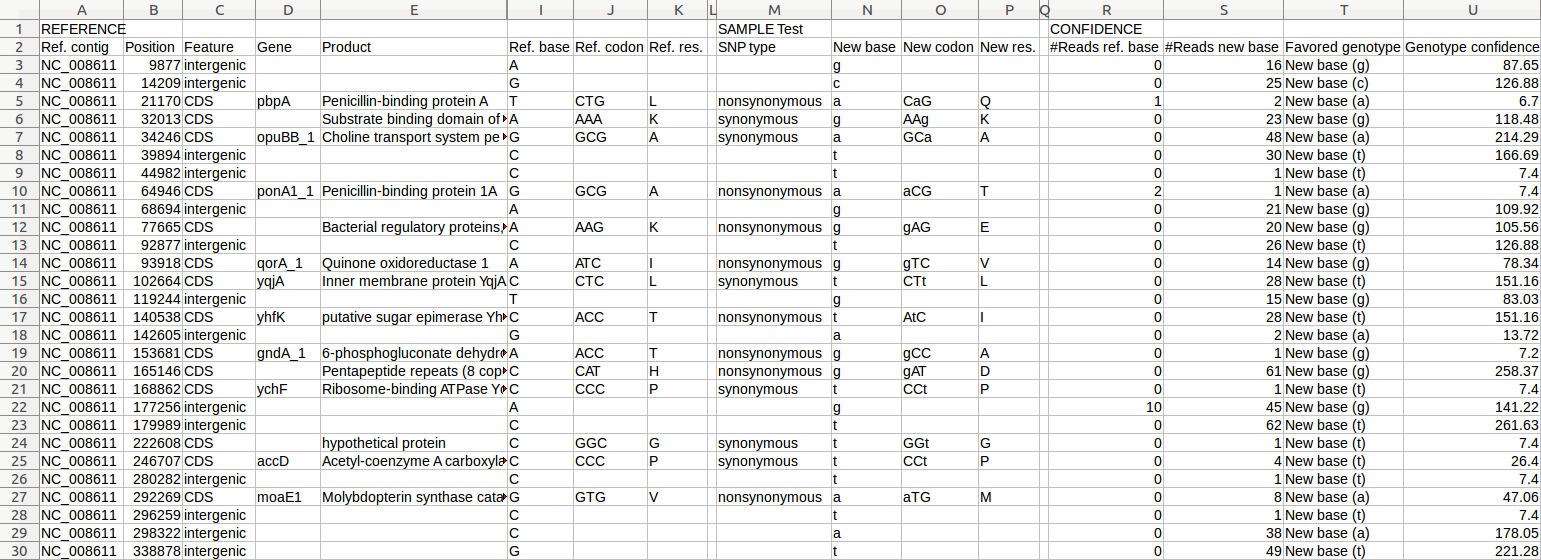
| TRAMS | SNP annotation | Functional annotation of SNP calls (from raw reads) | Examples |
When studying bacterial genomes, there are some vital processes aimed at the identification of the organism and its genomic characteristics, which can influence its pathogenic potential. The present module, composed of classification and functional analysis routines, makes IonGAP ideal for clinical bacteriology procedures involving bacterial genome sequencing, assembly and characterization. The applications which take part in this module and the results obtained from each one are detailed in the table below.
| Application | Process | Result | Result examples |
|---|---|---|---|
| BLAST, NCBI's 16S rRNA DB | Taxonomic classification | Tabular file containing 16S rRNA sequence alignments for each contig | Examples |
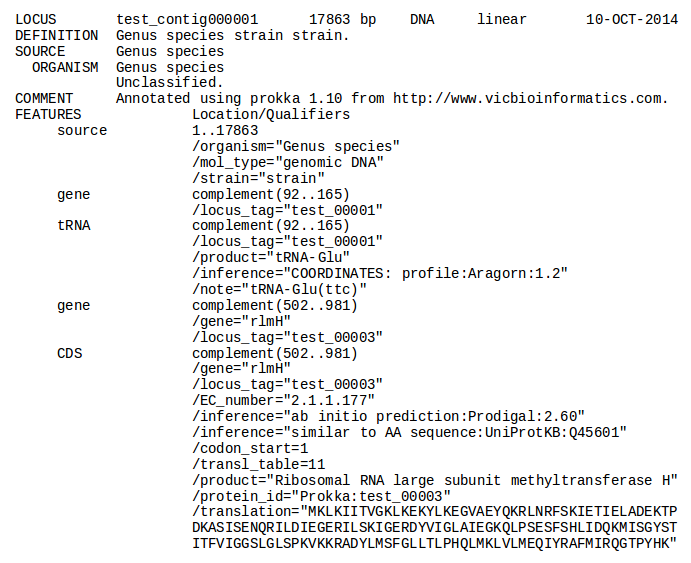
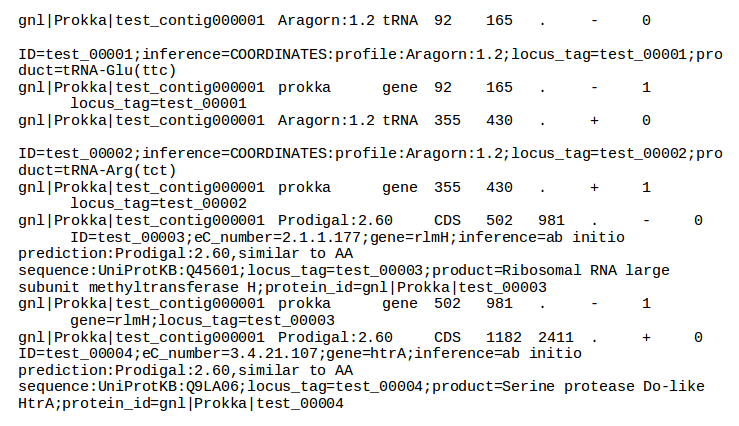
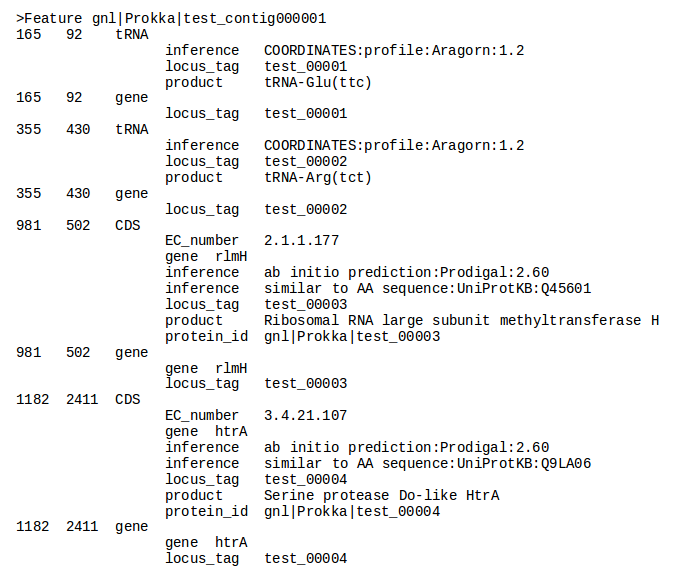
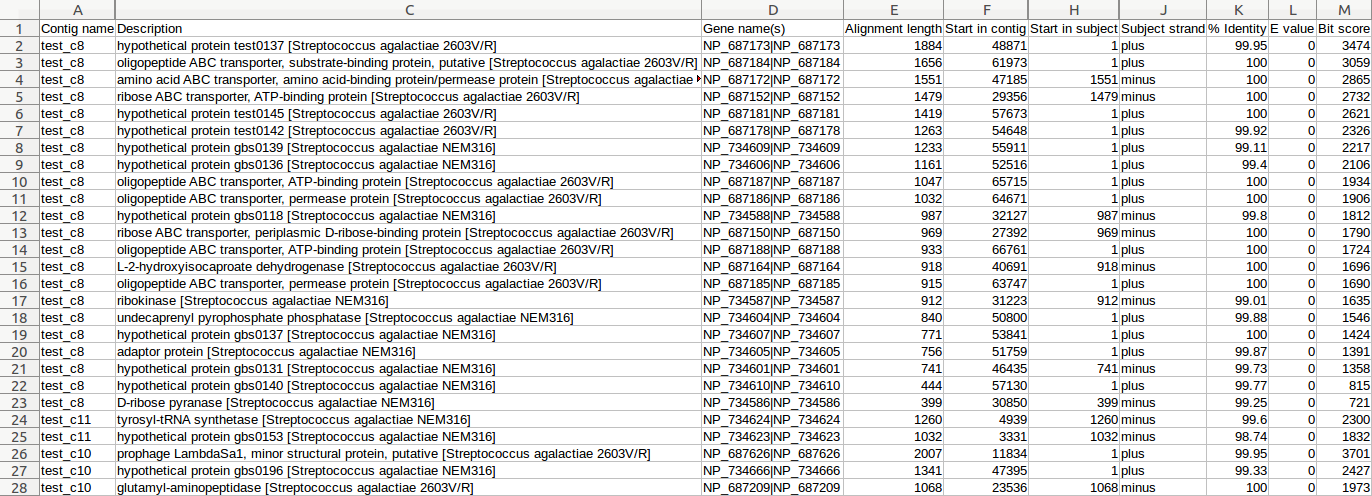
| Prokka | Genome annotation | Annotated contigs in several formats, annotated protein sequences | Examples |
| Torsten Seemann's mlst | Multilocus sequence typing | Identified ST and allele numbers, allele sequences | Examples |
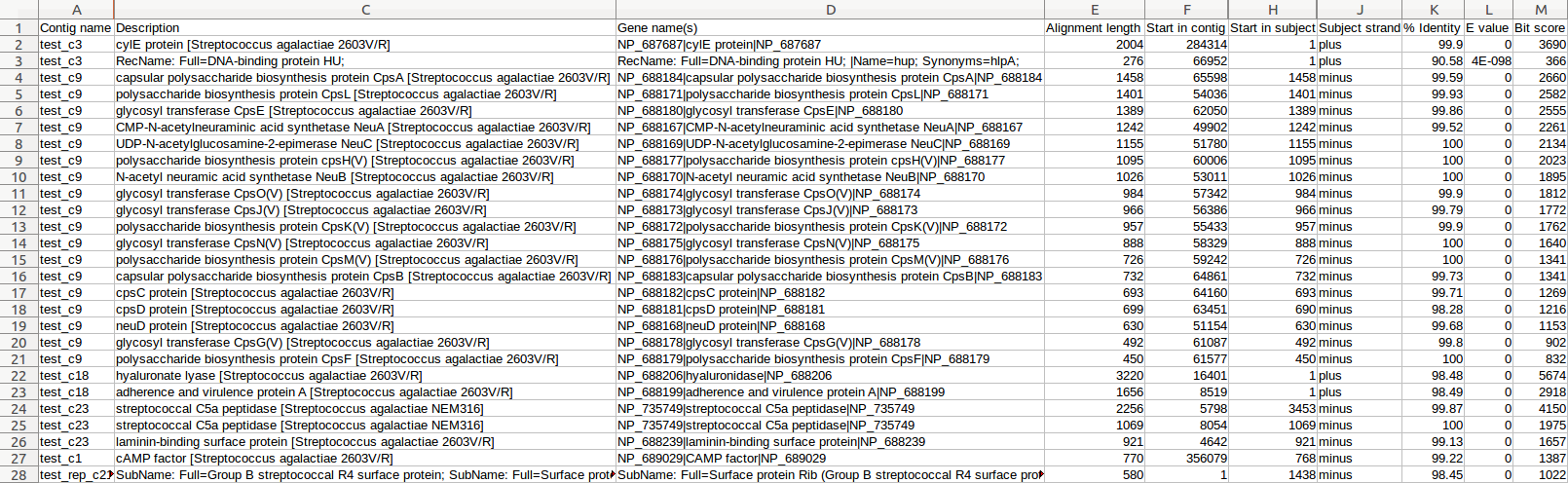
| BLAST, NCBI's Plamids DB | Identification of plamids | Tabular file containing plasmid sequence alignments for each contig | Examples |
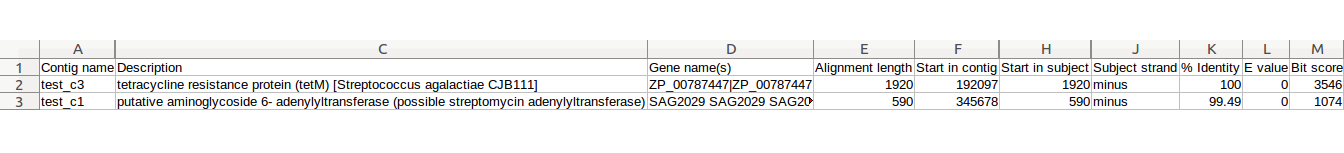
| BLAST, MvirDB | Identification of virulence factors | Tabular files containing sequence alignments of antibiotic resistance genes, virulence proteins and pathogenicity islands for each contig | Examples |
For more information about the pipeline results and how to interpret them, please consult the User Manual.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}